 |
ForceBalance
1.7.4
Automated optimization of force fields and empirical potentials
|
|
ForceBalance
1.7.4
Automated optimization of force fields and empirical potentials
|
The documentation for ForceBalance exists in two forms: a web page and a PDF manual. They contain equivalent content. The newest versions of the software and documentation, along with relevant literature, can be found on the SimTK website.
Users of the program should read the Introduction, Installation, Usage, and Tutorial sections on the main page.
Developers and contributors should read the Introduction chapter, including the Program Layout and Creating Documentation sections. The API documentation, which describes all of the modules, classes and functions in the program, is intended as a reference for contributors who are writing code.
ForceBalance is a work in progress; using the program is nontrivial and many features are still being actively developed. Thus, users and developers are highly encouraged to contact me through the SimTK website, either by sending me email or posting to the public forum, in order to get things up and running.
Thanks!
Lee-Ping Wang
Welcome to ForceBalance! :)
This is a theoretical and computational chemistry program primarily developed by Lee-Ping Wang and the Wang research group at UC Davis. The full list of people who made this project possible are given in the Credits.
The function of ForceBalance is automatic force field optimization. Here I will provide some background, which for the sake of brevity and readability will lack precision and details. In the future, this documentation will include literature citations which will guide further reading.
In theoretical and computational chemistry, there are many methods for computing the potential energy of a collection of atoms and molecules given their positions in space. For a system of N particles, the potential energy surface (or potential for short) is a function of the 3N variables that specify the atomic coordinates. The potential is the foundation for many types of atomistic simulations, including molecular dynamics and Monte Carlo, which are used to simulate all sorts of chemical and biochemical processes ranging from protein folding and enzyme catalysis to reactions between small molecules in interstellar clouds.
The true potential is given by the energy eigenvalue of the time-independent Schrodinger's equation, but since the exact solution is intractable for virtually all systems of interest, approximate methods are used. Some are ab initio methods ('from first principles') since they are derived directly from approximating Schrodinger's equation; examples include the independent electron approximation (Hartree-Fock) and perturbation theory (MP2). However, most methods contain some tunable constants or empirical parameters which are carefully chosen to make the method as accurate as possible. Three examples: the widely used B3LYP approximation in density functional theory (DFT) contains three parameters, the semiempirical PM3 method has 10-20 parameters per chemical element, and classical force fields have hundreds to thousands of parameters. All such formulations require an accurate parameterization to properly describe reality.
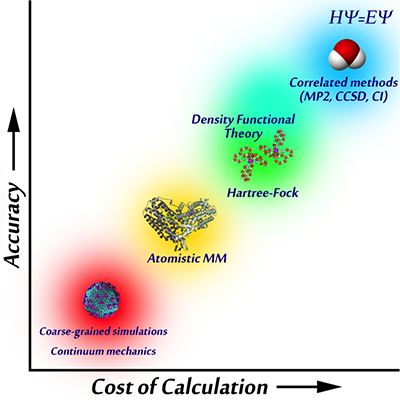
The main audience of ForceBalance is the scientific community that uses and develops classical force fields. These force fields do not use the Schrodinger's equation as a starting point; instead, the potential is entirely specified using elementary mathematical functions. Thus, the rigorous physical foundation is sacrificed but the computational cost is reduced by a factor of millions, enabling atomic-resolution simulations of large biomolecules on long timescales and allowing the study of problems like protein folding.
In classical force fields, relatively few parameters may be determined directly from experiment - for instance, a chemical bond may be described using a harmonic spring with the experimental bond length and vibrational frequency. More often there is no experimentally measurable counterpart to a parameter - for example, electrostatic interactions are often described as Coulomb interactions between pairs of atomic point "partial charges", but the fractional charge assigned to each atom has no rigorous experimental of theoretical definition. To complicate matters further, most molecular motions arise from a combination of interactions and are sensitive to many parameters at once - for example, the dihedral interaction term is intended to govern torsional motion about a bond, but these motions are modulated by the flexibility of the nearby bond and angle interactions as well as the nonbonded interactions on either side.
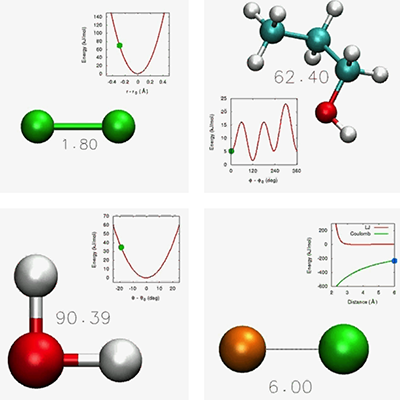
For all of these reasons, force field parameterization is difficult. In the current practice, parameters are often determined by fitting to results from other calculations (for example, restrained electrostatic potential fitting (RESP) for determining the partial charges) or chosen so that the simulation results match experimental measurements (for example, adjusting the partial charges on a solvent molecule to reproduce the bulk dielectric constant.) Published force fields have been modified by hand over decades to maximize their agreement with experimental observations (for example, adjusting some parameters in order to reproduce particular protein NMR structure) at the expense of reproducibility.
Given this background, I can make the following statement. The purpose of ForceBalance is to create force fields by applying a highly general and systematic process with explicitly specified input data and optimization methods, paving the way to higher accuracy and improved reproducibility.
At a high level, ForceBalance takes an empirical potential and a set of reference data as inputs, and tunes the parameters such that the simulations are able to reproduce the data as accurately as possible. Examples of reference data include energy and forces from high-level QM calculations, experimentally known molecular properties (e.g. polarizabilities and multipole moments), and experimentally measured bulk properties (e.g. density and dielectric constant).
ForceBalance presents the problem of potential optimization in a unified and easily extensible framework. Since there are many empirical potentials in theoretical chemistry and similarly many types of reference data, significant effort is taken to provide an infrastructure which allows a researcher to fit any type of potential to any type of reference data.
Conceptually, a set of reference data (usually a physical quantity of some kind), in combination with a method for computing the corresponding quantity with the force field, is called a target. For example:
Within a target, the accuracy of the force field can be optimized by tuning the parameters to minimize the difference between the computed and reference quantities. One or more targets can be combined to produce an aggregate objective function whose domain is the parameter space. This objective function, which typically depends on the parameters in a complex way, is minimized using nonlinear optimization algorithms. The result is a force field which minimizes the errors for all of the targets.
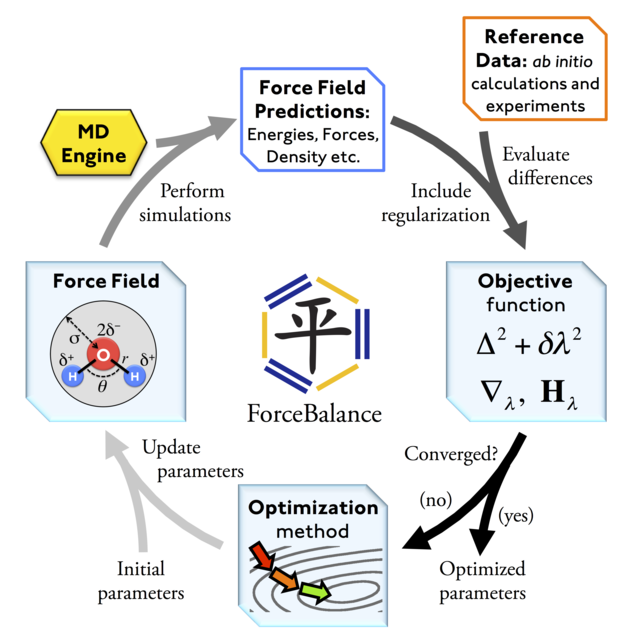
The problem is now split into three main components; the force field, the targets, and the optimization algorithm. ForceBalance uses this conceptual division to define three classes with minimal interdependence. Thus, if a researcher wishes to explore a new functional form, incorporate a new type of reference data or try a new optimization algorithm, he or she would only need to contribute to one branch of the program without having to restructure the entire code base.
The scientific problems and concepts that this program is based upon are further described in my Powerpoint presentations and publications, which can be found on the SimTK website.
The development of this code has been supported in part by the following grants and awards:
NIH Grant R01 AI130684-02 ACS-PRF 58158-DNI6 NIH Grant U54 GM072970 Open Force Field Consortium
 1.8.13
1.8.13